Our group has several active research themes in electrophilic catalysis, which are primarily concerned with the use of the Brønsted superacid bis-trifluoromethane sulfonimide Tf2NH and its Lewis superacid metal salt derivatives M(NTf2)n, but also include organocatalysis, gold chemistry and multicatalysis which all find utility in the chemistry of N-acyliminium ions and rapid construction of aza-heterocycles.
Brønsted and Lewis superacids :
We have recently investigated many aspects of catalytic N-acyliminium ion chemistry and thus developed efficient and general protocols which mainly use trifluoromethanesulfonamide-based catalysts [both Tf2NH and its metal complexes, notably Sn(NTf2)4] at a loading rarely exceeding 1 mol %.[7-9] Using common (O-acylated and O-alkyl) N,O-acetals, these superacids were indeed shown to be highly efficient pre-catalyst (Tf2NH) and catalyst (Sn(NTf2)4) for the amidoalkylation of various C-nucleophiles including silicon-based nucleophiles (silyl enol ethers, ketene silyl acetals, allyltrimethylsilane, TMSCN…), β-dicarbonyl nucleophiles and activated arenes.
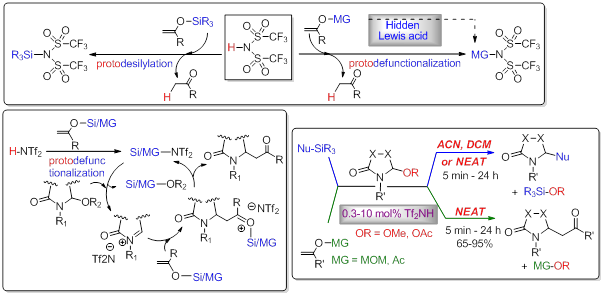
The high efficiency of the α-amidoalkylation reactions of N,O-acetals with silylated nucleophiles catalyzed by Tf2NH is based on the well acknowledged mechanistic paradigm that the Brønsted superacid is a pre-catalyst that triggers a silylative catalysis of the reaction due to an early protodesilylation of the silicon-based nucleophile. Based on these considerations, a mechanism-guided designed strategy has allowed us to extend these organocatalytic amidoalkylations to the use of enol ethers bearing cationic carbon migrating groups (MG = MOM, acyl.... in lieu of the trialkyklsilyl subunit), which behave as hidden organic Lewis acids. This methodology proceeds in an extremely efficient manner, typically requiring 1-2 mol% Tf2NH in neat conditions, short reaction times, and affording the Mannich adducts in high yields.
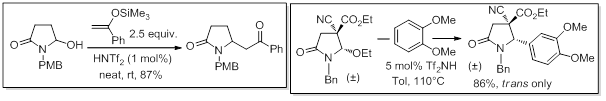
The high acidity strength of these catalysts also enables high performance in the challenging alkylation of poorly reactive N,O-acetals. This includes, as representative examples, the simplest but less reactive hydroxy hemi aminals (hydroxy lactames and related structures), and N,O-acetals sterically and electronically deactivated by two adjacent electron-withdrawing groups (CN,CO2R or CO2R,CO2R). The latter are readily available from a methodology based on formal [3+2] cycloadditions of α-bromoacetamides developed in our laboratory (see theme domino and tandem reactions).
Based on this unique acidic capacity of triflimide-based reagents, we have recently developed the first catalytic α-amidoalkylation of simple ketones, which is one the most notable achievements in this theme. Sn(NTf2)4 is the optimal catalyst for this transformation which has been developed by the way of two complementary variants, typically using only 1 mol% of loading. One is atom-economical and involves hemi aminals (hydroxy lactames) which are readily alkylated at elevated temperature (≥60°C), while the alternative method typically proceeds at room temperature and generally requires more active N,O-acetals such as acetoxy lactams.
Enantioselective Organocatalysis :
We have also recently initiated a program aimed at developing catalytic enantioselective alkylations of N-acyliminium ions using chiral Brønsted acid catalysts (Asymmetric Counterion Directed Catalysis strategy). Basically, the same categories of reactions as the ones discussed above are being studied.
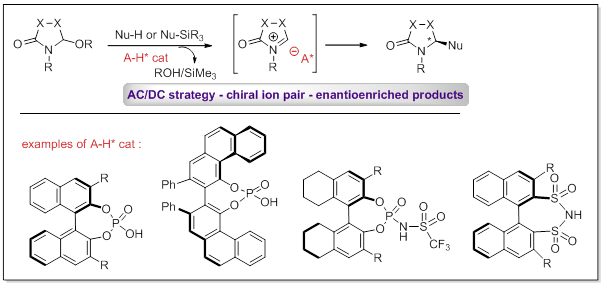
Gold catalysis :
Very recently the group has become attracted to use gold catalysis in the context of this chemistry. Notably, our usual superacid catalysts having been unable to efficiently catalyze the alkynylation of N-acyliminiums ions starting from N,O-acetals and diverse sources of alkynyl nucleophiles, the use of gold chemistry has proven to be an excellent choice to solve this problem. The first catalytic alkynylation of N,O-acetals exploiting an unprecedented combination of TMS-alkynes and gold(I) salt catalysts having triflate or triflimidate counterions, has thus been developed. The reaction is generally an efficient process that affords the desired products in high yields and with large scope. The reaction mechanism is reminiscent to the one presented above for the Tf2NH-catalyzed α-amidoalkylation of silyl enol ethers, with the difference that the protodesilylation is replaced by an aurodesilylation, which means that the sacrificial catalytic amount of silicon-based nucleophile (released in the form of ketone) in the above approach is here translated into a gold acetylide, which, as the key nucleophile of the process, is an active species in our catalytic cycle.
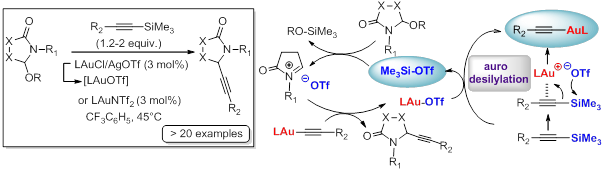
Multicatalysis :
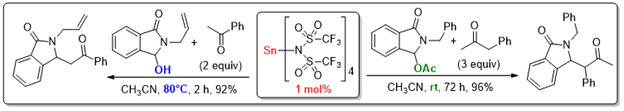
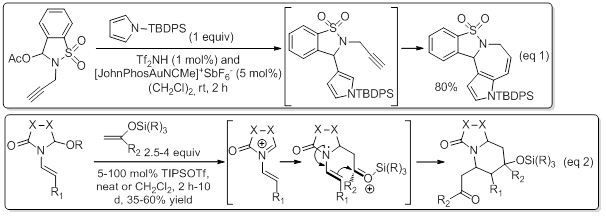
In general, our research is largely inspired by the respect of some important principles of sustainable chemistry such as eco-compatibility, atom and step economy. This is reflected by development of multicatalytic strategies, where the catalytic process involving N-acyliminium ion is combined with one or more other catalytic transformations, allowing quick and easy access (one step) to relatively sophisticated (poly)heterocyclic structures from trivial starting materials. Two representative examples of such multicatalytic cascades are : 1- a Tf2NH-catalyzed intermolecular Friedel-Crafts amidoalkylation/gold-catalyzed intramolecular hydroarylation sequence, that provides expeditious access to diverse polyazaheterocyclic structures (see eq 1), while illustrating another use of gold catalysis in the context of N-acyliminium ion chemistry. 2- A cascade sequence Mannich type amidoalkylation-Prins cyclization, i.e. formally a [4 + 2] cycloaddition event, catalyzed by TIPSOTf which provides an expeditious access to highly functionalized indolizidine frameworks.
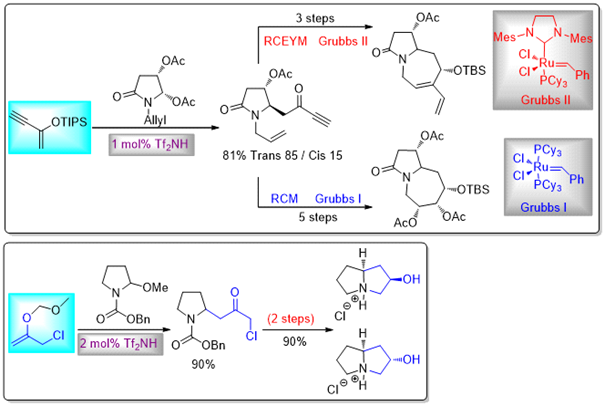
Related chemistry where the catalytic Mannich reaction was used as a key step in short synthetic routes towards to (poly)hydroxylated bicyclic iminosugars have also been developed. This has been achieved by using dienoxysilanes or ynenoxysilanes as nucleophilic partners in the amidoalkylation process, in conjunction with a subsequent ring-closing metathesis event, or by using the dipolarophilic MOM enol ether of chloroacetone, which can subsequently afford access to pyrrolizidines through intramolecular chloride displacement.